Paul K. Drain, Andre B. Gvozden, Melanie Hoppers, Stephen Young, Michalina Montano
In an analysis of 257 subjects presenting with symptoms of COVID-19, the LumiraDx SARS-CoV-2 Ag 12-minute nasal swab test had 97.6% sensitivity and 96.6% specificity within 12 days of symptom onset compared to RT-PCR. This period represents the period of infectivity*. All (100%) samples detected within 33 RT-PCR cycles were also identified using the antigen test.
Methods
A prospective cohort study was conducted to evaluate the LumiraDx SARS-CoV-2 antigen Test among children and adults who presented for COVID-19 testing. The study was conducted at six sites across the United States and United Kingdom in which minimally trained operators tested specimens at the point of care. The clinical study received ethical approval and all participants provided informed consent (NCT 04557046). After obtaining sociodemographic and clinical data, two anterior nasal swabs (Copan FLOQ swab) were collected from each participant. One swab was placed into 0.7 ml of proprietary extraction buffer for the LumiraDx SARS-CoV-2 antigen test, and the second swab was placed into 3 ml of viral transport media for RT-PCR testing using a Roche Cobas 6800 platform (Roche Diagnostics, Indianapolis, IN, USA). We evaluated diagnostic performance and stratified results by age, gender, days since symptom onset, and RT-PCR cycle threshold (Ct) values, and used a Wilson 2-sided analysis for 95% confidence intervals (CI).
Results
Among 257 people enrolled, ages ranged from 0-90 years and 142 (55%) were female. 159 (62%) people experienced symptoms of COVID-19 with an average duration of 4 days at the time of testing. Based on RT-PCR testing, 83 nasal swabs were positive for SARS-CoV-2, giving an estimated infection prevalence of 32.3% (95% CI 26.9-38.2%).
As expected, the RT-PCR Ct values increased with more days since the onset of symptom. Overall, the LumiraDx SARS-CoV-2 antigen Test had a sensitivity of 97.6% (95% CI 91.6-99.3%) and specificity of 96.6% (95% CI 92.7-98.4%) up to 12 days since symptom onset. There were no appreciable differences when results were stratified by age or gender. The SARSCoV-2 antigen Test was highly sensitive up to a Ct value of 33 cycles, and two participants with a false negative antigen test both had RT-PCR Ct values ≥33 cycles (100% sensitive for RT-PCR cycle threshold <33 cycles). When restricted to people testing within 10 days of symptom onset, which likely correlates to a period of viral culturability, the diagnostic sensitivity was 98.7% (95% CI 93.0-99.8%) .
Conclusion
In summary, the LumiraDx SARS-CoV-2 antigen test demonstrated high sensitivity when used to aid diagnosis of COVID-19 at the clinical point of care. The rapid test was highly sensitive for people with RT-PCR Ct values of <33 cycles within a period of 12 days since the onset of COVID-19 symptoms. These performance characteristics may correlate well with reported infective SARS-CoV-2 viral load and window of infectivity. The test achieved a lower Limit of Detection (LoD) and higher diagnostic sensitivity than other Point of Care (POC) tests and provided fast results (in under 12 minutes), in a convenient, easy to use point of care test format, with capacity to transfer data to electronic health records and surveillance systems.
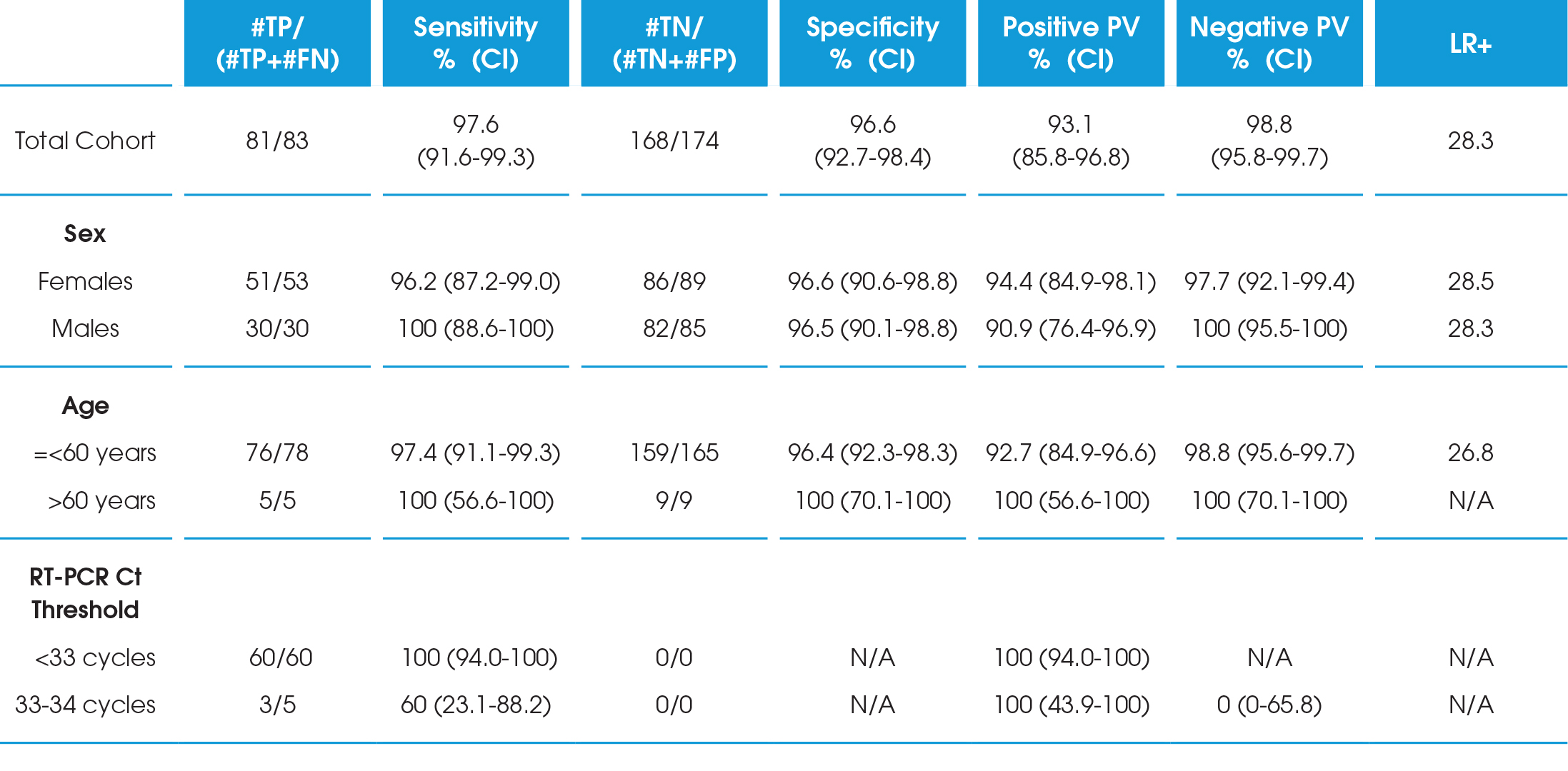
CI=Confidence Interval; FN=false negative; FP=false positive; PV=Predictive Value; TN=true negative; TP=true positive; LR+=Likelihood Ratio Positive.
*R. Wölfel, V.M. Corman, W. Guggemos, M. Seilmaier, S. Zange, M.A. Müller, D. Niemeyer, T.C. Jones, P. Vollmar, C. Rothe, M. Hoelscher, T. Bleicker, S. Brünink, J. Schneider, R. Ehmann, K. Zwirglmaier, C. Drosten, C. Wendtner, Virological assessment of hospitalized patients with COVID-2019. Nature. 581(7809), 465-9 (2020) DOI: 10.1038/s41586-020-2196-x
For use by healthcare professionals only. Products not available in all countries or regions. Available in the USA under FDA Emergency Use Authorization. In the USA, this test has not been FDA cleared or approved; this test has been authorized by FDA under an EUA for use by authorized laboratories; use by laboratories certified under the CLIA, 42 U.S.C. §263a, that meet requirements to perform moderate, high or waived complexity tests. This test is authorized for use at the Point of Care (POC), i.e., in patient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation. This test has been authorized only for the detection of proteins from SARS-CoV-2, not for any other viruses or pathogens. In the USA, this test is only authorized for the duration of the declaration that circumstances exist justifying the authorization of emergency use of in vitro diagnostics for detection and/or diagnosis of the virus that causes COVID-19 under Section 564(b)(1) of the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.